Орфанные заболевания – это редкие и потому зачастую малоизученные болезни. Многие из них – генетические – а значит, лечить их очень сложно и дорого. Дети с орфанными заболеваниями редко доживают до совершеннолетия. Болезнь может проявиться и у взрослого.
Редкие болезни сложно изучать, и еще сложнее разрабатывать лечение от них. Поэтому лекарства, если они существуют, очень дороги. Их нельзя делать массово, а производителям необходимо, чтобы исследования, разработка и производство окупались.
По разным оценкам, в мире существует 5-7 тысяч орфанных заболеваний. Составить их полный перечень невозможно. Во-первых, нет четкого определения, что такое редкие заболевания. Во-вторых, наверняка существуют еще неизученные состояния.
Мы составили перечень из 12 орфанных заболеваний. О некоторых из них вы наверняка знаете, а о других впервые прочитаете в нашей статье.
Что такое орфанные заболевания
Определения у редких заболеваний нет. В разных странах орфанными заболеваниями называют болезни с разной частотой.

| Страна | Частота встречаемости заболевания |
| Россия | 1/10000 |
| США | 1/1500 |
| Япония | 1/2500 |
| Страны Евросоюза | Менее 1/2000 |
Европейская комиссия по общественному здравоохранению называет орфанными заболеваниями (rare diseases, orphan diseases) опасные для жизни или хронические болезни, изучать и бороться с которыми можно только совместными усилиями.
А Европейская организация редких заболеваний EURORDIS относит к орфанным и редкие, и забытые болезни. Забытыми болезнями называют инфекции или паразитарные инвазии, которыми болеют в основном в бедных странах. От них есть средства, но у больных обычно нет ресурсов на лечение.
Дать определение орфанным заболеваниям сложно и потому, что в разных странах и среди разных групп людей частота болезней различается. Иногда это зависит от возбудителя – к примеру, в России мало кто из врачей ожидает встретить больного сонной болезнью или дирофиляриозом .
В случае генетических болезней частая причина их большего распространения среди каких-либо людей – эффект основателя.
Когда небольшая группа людей колонизует новые земли или живет в изоляции, генетическое разнообразие у них ниже, и некоторые гены встречаются чаще, чем в более разнородной группе. Это может стать причиной, по которым генетическая болезнь, редкая во всем мире, будет чаще поражать людей определенного происхождения.
Перечень орфанных заболеваний
В России орфанными считаются 262 болезни. Все они включены в Перечень редких заболеваний Минздрава. По официальной статистике, суммарное число больных ими на февраль 2020 года – около 15800 человек. Многие орфанные заболевания приводят к инвалидности в детском возрасте. Таким детям помогает Фонд поддержки детей.
Наша подборка из 12 орфанных заболеваний включает самые опасные, социально значимые и необычные состояния.
Зигомикоз
Зигомикоз, или мукормикоз – единственное орфанное заболевание в нашем списке, которым можно заразиться, хотя и не от других людей. Его вызывают плесневые грибы – мукор.
Плесени живут везде: в почве, компостных кучах, на продуктах. Их споры переносятся по воздуху на большие расстояния, так что каким бы чистым ни был дом, в нем почти наверняка есть споры грибов.
Зигомикоз обычно возникает у людей с иммунодефицитом, независимо от его причин. Его наблюдали у больных диабетом или онкологическими заболеваниями, ВИЧ-инфицированных, после приема лекарств, подавляющих иммунитет, при трансплантации органов.
Возможны несколько вариантов инфекции:
- Кожная – когда мукор поражает кожу и подкожные ткани. Рана опухает, повышается температура. Кожа вокруг повреждения чернеет.
- Легочная – возникает при вдыхании спор мукора. Плесневой гриб поражает легкие и вызывает кашель, повышение температуры и нарушения дыхания. Этой формой зигомикоза нередко страдают рабочие, ответственные за замену фильтров кондиционеров. Встречается она и у археологов.
- Если проглотить споры мукора, например, с плесневелыми продуктами, может развиться желудочно-кишечная форма зигомикоза. Она чаще возникает у новорожденных детей, поскольку их кишечная микрофлора еще плохо противостоит бактериям и грибам извне.
- При риноцеребральной форме зигомикоза мукор прорастает на лице и повреждает глаза, нёбо, нос, лицевые кости. Пораженные ткани чернеют. Если не лечить эту болезнь, гифы гриба проникнут в мозг. При этом возможны нарушения восприятия, галлюцинации, коматозное состояние. Риноцеребральная форма зигомикоза нередко смертельна.
- Диссеминированная – с поражением нескольких органов и тканей тела. Как правило, смертельна, поскольку зигомикоз быстро развивается и его сложно диагностировать.
У людей без нарушений иммунитета из перечисленных форм болезни наблюдали только кожный зигомикоз.

Споры мукора могут попасть в загрязненную землей рану или в повреждения от шипов растений. Гриб поражает окружающие рану ткани: кожу, подкожную клетчатку, жир. Иногда гифы дорастают до мышц и костей. Если мукор попадет в кровеносную систему, возникнет диссеминированная форма зигомикоза.
Мукор вызывает некроз тканей – их гибель. Поэтому пораженные этим грибом ткани удаляют хирургически. Часто после зигомикоза требуются пластические операции, чтобы восстановить внешность.
Чтобы уничтожить мукор, используют противогрибковые препараты. Однако поскольку зигомикоз протекает быстро, а отличить его от других болезней с похожим течением сложно, правильное лечение не всегда успевают даже начать.
Мукормикозом болеют не только люди, но и животные, например, ящерицы. У них он возникает, если овощи и фрукты в террариуме редко заменять и позволить им плесневеть.
Альбинизм
Альбинизм – это отсутствие меланина. Этот пигмент придает темную окраску волосам или шерсти, коже, глазам, защищает нас от ультрафиолета и участвует в развитии глаз и зрительных нервов.
Альбиносы легко обгорают на солнце, и у них часто снижено зрение из-за светобоязни, нарушений работы зрительных нервов, повреждений сетчатки или по другим причинам.

У альбиносов меланин может отсутствовать только в глазах (глазной альбинизм) или и в глазах, и в коже (кожно-глазной альбинизм). При этом в зависимости от формы альбинизма кожа у них может быть как белой, так и желтоватой или нормально окрашенной (при глазном альбинизме).
Волосы у альбиносов белые, желтые или рыжие. Цвет глаз также зависит от формы альбинизма. Чаще всего альбиносы голубоглазые или зеленоглазые, но у них бывают и карие глаза. Причина такого разнообразия в том, что цвет волос и кожи определяется не только меланином. Цвет глаз определяют не только пигменты, но и плотность волокон белка коллагена. От меланина этот фактор не зависит.

При альбинизме чаще всего нарушается работа гена, отвечающего за производство белка тирозиназы. Этот фермент участвует в синтезе меланина.
В результате разных мутаций тирозиназа отключается полностью или частично или работает только при температуре ниже 37°С. Последнее называют температуро-чувствительным альбинизмом. При нем у новорожденных детей меланин отсутствует, но с возрастом большая часть кожи приобретает нормальный цвет, а волосы темнеют. Но области под мышками у них белые или желтоватые.
Некоторые формы альбинизма связаны с нарушениями работы других систем. Например, при синдроме Германски-Пудлака снижается свертываемость крови, а при синдроме Чедиака-Хигаси нарушена работа иммунной системы.
Методов лечения альбинизма в настоящее время не существует. Им рекомендуют использовать очки и кремы, чтобы защитить глаза и кожу от ультрафиолета. Альбиносам следует регулярно посещать окулиста и дерматолога, чтобы не допустить развития нарушений зрения и новообразований кожи.
Некоторые лекарства способствуют синтезу меланина у альбиносов. Одно из таких исследований закончили в 2019 году. Публикацию можно прочитать по ссылке: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6413781/. Однако выборки из 5 пациентов недостаточно, чтобы утверждать что-либо об эффективности такого метода лечения альбинизма.
В Европе и России альбиносы встречаются редко, и к ним относятся спокойно. В странах Африки альбиносов убивают, чтобы затем использовать их части тела для «колдовских зелий» или преследуют из-за мифа о том, что альбиносы способны излечивать ВИЧ.
Врожденный ихтиоз
Название «ихтиоз» – однокоренное со словом «ихтис», «рыба». При этих редких заболеваниях кожа сильно шелушится и напоминает рыбью чешую.
Это состояние может возникать из-за лекарств или других болезней, например, гипотиреоза. К орфанным заболеваниям относят врожденные ихтиозы. Их причина – различные дефекты генов, связанных с выработкой кератинов. Из этих белков состоят волосы и ногти людей, шерсть и когти животных, перья птиц.
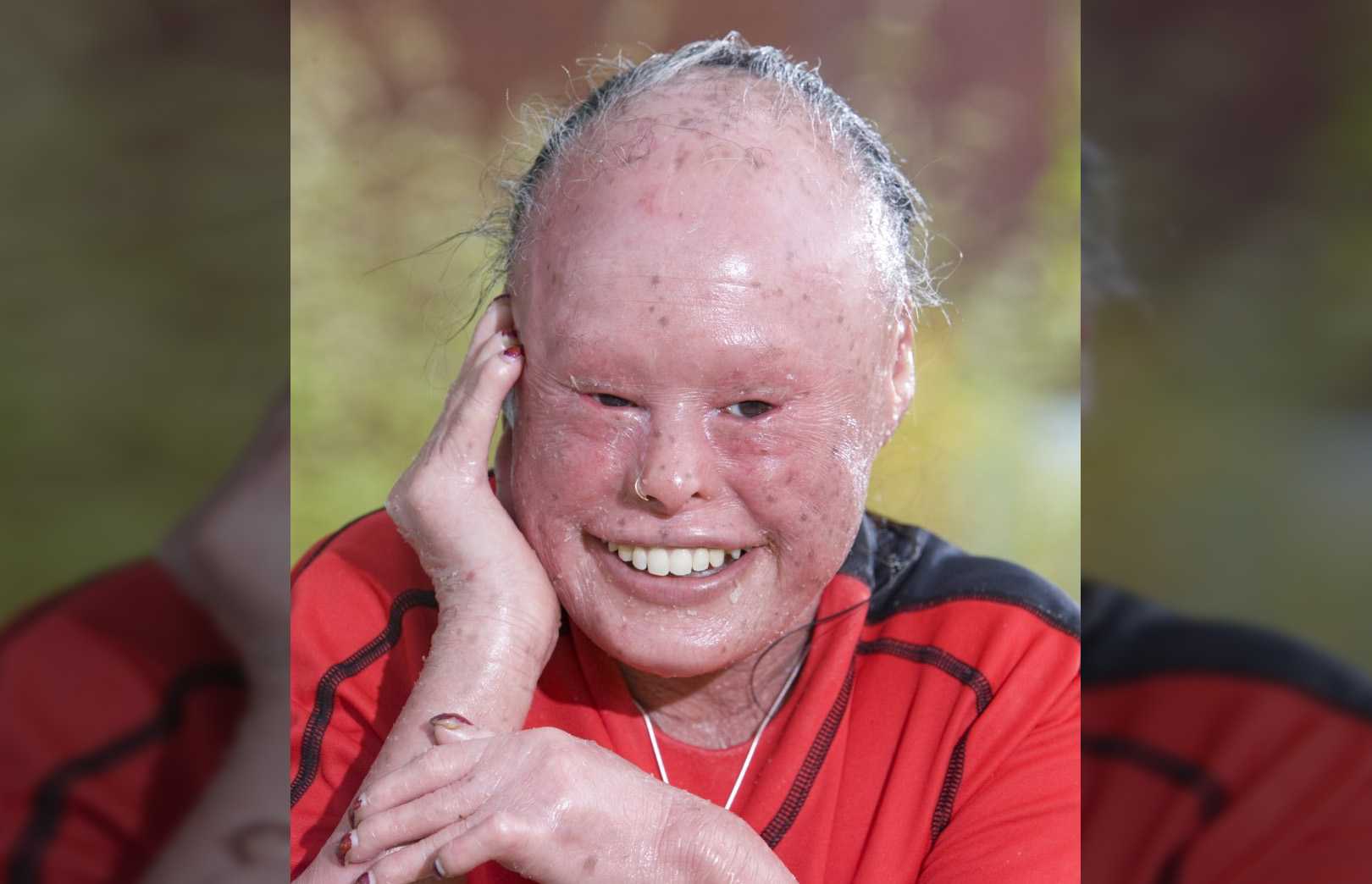
Возможны несколько разных причин ихтиозов:
- Избыток кератина;
- Слишком прочная связь кератиновых чешуек друг с другом;
- Неправильное складывание и нарушение функций кератина.
Все это приводит к изменению вида кожи, ее сухости и повышенной потере воды.
Вылечить врожденный ихтиоз в настоящее время невозможно. При этих болезнях рекомендуют использовать средства, увлажняющие кожу и препараты для разрушения кератина. Довольно часто врачи назначают при ихтиозах ретиноиды – различные формы витамина А.
Эти вещества при длительном приеме накапливаются в организме и вызывают отравление. Поэтому за состоянием пациентов необходимо регулярно наблюдать.
Муковисцидоз
Муковисцидоз, или фиброзный кистоз – результат мутаций гена, отвечающего за один из белков, переносящих ионы хлора из клетки наружу.
Этот процесс особенно важен для клеток слизистых оболочек легких, пищеварительной системы, протоков поджелудочной, слюнных, потовых и половых желез. При муковисцидозе их клетки разрушаются, и в легких, пищеварительном тракте и железах накапливается густая слизь.
В результате возникают хронические бронхиты и пневмонии, нарушения пищеварения, повреждения поджелудочной железы и печени. Муковисцидоз обычно выявляют при скрининге новорожденных. Живут больные муковисцидозом, в зависимости от времени диагностики и качества лечения, до 20-40 лет.
Большинство методов лечения муковисцидоза – симптоматические. Больные муковисцидозом принимают бронхорасширяющие препараты, лекарства для разжижжения слизи и противовоспалительные. Им необходимо пожизненно придерживаться диеты с ограниченным содержанием жиров и высоким – белков, соли и жидкости. Иногда при тяжелых поражениях легких больным трансплантируют донорские органы.
Муковисцидоз могут вызывать различные мутации. Некоторые из них препятствуют синтезу или правильному складыванию белка, при других же белок плохо работает.
Для лечения муковисцидоза при некоторых мутациях используют препарат «Калидеко» (действующее вещество – ивакафтор) или его аналоги. Это вещество поддерживает белок в открытом состоянии. В результате ионы хлора выводятся из клеток, клетки не погибают, и работа внутренних органов нормализуется. Ивакафтор и его аналоги помогают не при всех формах муковисцидоза и не лечат саму причину нарушений. Но они значительно улучшают жизнь больных.
Амилоидоз
Амилоидоз – нарушение обмена веществ, при котором в тканях откладываются соединения белков с углеводами – амилоиды.
В зависимости от того, где откладываются амилоиды, возникают нарушения работы разных органов и систем. Чаще всего страдают почки, на втором месте – органы желудочно-кишечного тракта и селезенка.
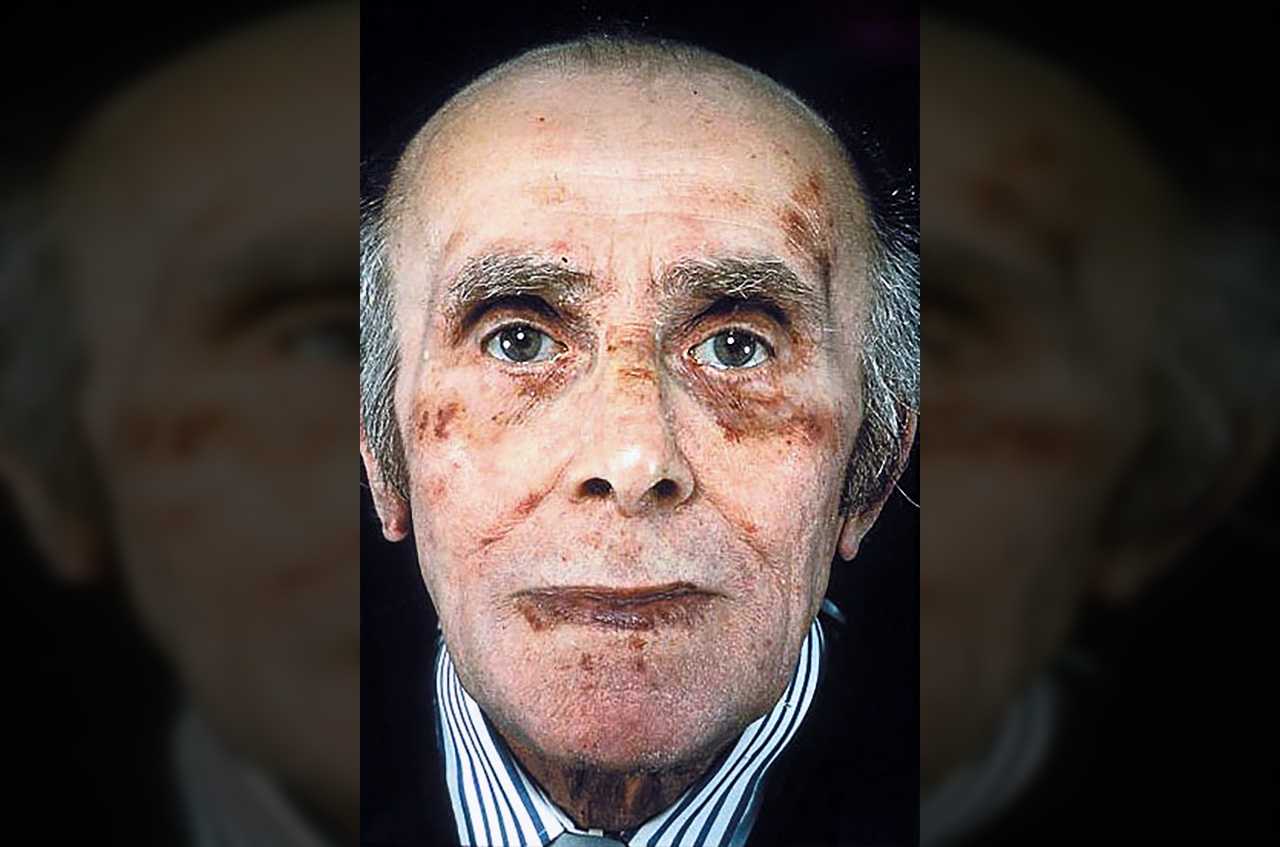
Некоторые формы амилоидоза обусловлены генетически и вызваны мутациями. К таким относится, например, средиземноморская перемежающаяся лихорадка или амилоидоз финского типа. Наследственные формы амилоидоза часто привязаны к определенным странам.
Амилоидоз может возникать в результате гемодиализа – очистки крови от продуктов обмена веществ на аппарате «искусственная почка». При этом в организме накапливаются определенные белки, которые в норме удаляются почками.
Иногда амилоидоз возникает с возрастом, при нарушениях работы печени или в из-за ревматоидного артрита, туберкулеза или других болезней.
Для лечения амилоидоза используют препараты, подавляющие работу иммунной системы, в том числе, колхицин и преднизолон. Иногда больным пересаживают донорские органы.
Если амилоидоз вызван избыточной выработкой какого-либо белка, можно снизить активность его синтеза и, следовательно, замедлить развитие болезни. Такое лечение применяют против наследственного транстиретинового амилоидоза, при котором организм производит слишком много белка транстиретина. Этот белок участвует в транспорте ретинола (витамина А) и тироксина (одного из гормонов щитовидной железы).
Препараты инотерсен и патисиран способствуют разрушению РНК, кодирующей транстиретин и, следовательно, снижают его выработку печенью.
Фенилкетонурия
Фенилкетонурия – наследственное заболевание, при котором организм вырабатывает слишком мало фермента фенилаланингидроксилазы или совсем не вырабатывает его.
Фенилаланингидроксилаза расщепляет аминокислоту фенилаланин в печени. В результате фенилаланин разрушается иными путями и превращается в ядовитые вещества, которые накапливаются в организме.
Это приводит к судорогам и другим нарушениям работы мышц и нервной системы, изменению запаха пота и мочи, нехватке меланина и, без лечения – к тяжелой умственной отсталости.

Фенилкетонурию диагностируют у новорожденных не младше 10 дней или в 2-4 месяца. Поэтому им вовремя назначают лечение и диету без фенилаланина. Если соблюдать ее до совершеннолетия или пожизненно, ребенок развивается нормально, и его интеллект не страдает.
Фенилаланин содержится в большинстве белков. Поэтому при фенилкетонурии детям назначают диету без мяса, рыбы, молочных и многих растительных продуктов. Им нельзя пить напитки и есть сладости с фенилаланином, поэтому продукты с этой аминокислотой маркируют.
Генная терапия фенилкетонурии и лечение на основе введения в организм аналогов фенилаланингидроксилазы находятся в разработке. Публикацию о генной терапии этого заболевания можно прочитать по ссылке: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6763965/
Рецессивный поликистоз почек
При поликистозах почек в их тканях появляются полости – кисты. Переродившиеся таким образом почки не могут выводить из организма продукты обмена веществ, что приводит к смерти.
Поликистозы почек – наследственные заболевания. Для развития доминантного поликистоза почек достаточно одной поврежденной копии любого из трех генов, кодирующих полицистины 1, 2 или 3. Без этих белков почки работают неправильно: их клетки делятся слишком активно и образуют кисты.
В мире около 12 миллионов человек больны доминантным поликистозом. Чаще всего болезнь проявляется после 30 или даже после 50 лет.
Чтобы развился рецессивный поликистоз почек, необходимы две сломанных копии гена, кодирующего фиброцистин. Это – редкое совпадение, поэтому рецессивный поликистоз почек относят к орфанным заболеваниям.
Болезнь можно диагностировать внутриутробно. При рецессивном поликистозе почки иногда недоразвиваются, из-за чего 30% больных умирают вскоре после рождения. У остальных в почках образуются кисты.
В настоящее время не существует методов лечения рецессивного поликистоза почек. Больным необходимо соблюдать строгую диету с низким содержанием белка и без кофеина, а также контролировать артериальное давление и избегать лекарств, способных повредить почки.
Синдром де ла Туретта
В отличие от многих орфанных болезней, синдром Туретта не смертелен.
Синдром Туретта затрагивает только центральную нервную систему, причем мышление при этом не страдает. Нарушается только способность человека контролировать свои движения и слова. Это называется моторными и вербальными тиками.
Вопреки распространенному мнению, вербальные тики – не всегда ругательства (копролалия). Чаще всего тики – это частое моргание, пожатия плечами или кашель.
Синдром Туретта – генетическое заболевание, но науке пока неизвестно, мутации какого гена или генов его вызывают и как на его развитие влияют факторы окружающей среды.
При легких формах синдрома Туретта лекарства не требуются. При более тяжелых пациентам иногда назначают антипсихотики против тиков или антидепрессанты и литий от проблем с концентрацией внимания.
Эритропоэтическая порфирия
Порфириями называют болезни, при которых в организме накапливаются порфириногены. Это предшественники гема, который входит в состав гемоглобина. На свету они превращаются в порфирины, которые, взаимодействуя с кислородом, повреждают клетки организма.
Особенно страдают при этом кожа и нервная система. При порфириях возникают нарушения работы пищеварительной системы, боли в животе и груди. Частота сердцебиения и кровяное давление повышаются. Моча больных окрашена в красный цвет. Этот симптом позволяет быстро выявить болезнь.

Слово «эритропоэтическая» означает, что болезнь вызвана неправильной работой костного мозга. Более распространенные печеночные порфирии вызваны нарушениями работы печени.
Эритропоэтическая порфирия, или болезнь Гюнтера – очень редкое наследственное заболевание. Для его развития необходимы две поврежденные копии гена – по одной от каждого из родителей.
При болезни Гюнтера у людей на теле и лице много волос – иногда создается впечатление, что человек покрыт шерстью. Это называют гипертрихозом. Поскольку порфирины накапливаются в зубной эмали, зубы у больных порфирией красноватые. Свет вызывает у больных тяжелые ожоги.
Обычно болезнь Гюнтера выявляют у новорожденных и позднее – у детей 4-5 лет. Лечения от нее не существует. Больным рекомендуется избегать солнечного света и носить одежду, которая защитит их от воздействия ультрафиолета. Иногда больным пересаживают костный мозг здоровых людей.
Болезнь Виллебранда
Болезнь Виллебранда проявлениями похожа на гемофилию. При обеих этих болезнях кровь плохо свертывается, и любое кровотечение опасно для жизни.
В отличие от гемофилии, болезнь Виллебранда может возникать и у мужчин, и у женщин. Кроме того, при этом заболевании стенки сосудов слишком легко растягиваются и пропускают кровь. Поэтому при болезни Виллебранда кровотечения могут возникать спонтанно, без повреждений.
Кровоточат слизистые носа и рта, а также внутренние органы. Кровь изливается под кожу, в полости органов и суставов. При травмах или приеме лекарств, снижающих свертываемость крови возможны мозговые кровоизлияния.
Самая распространенная форма болезни Виллебранда наследуется доминантно. Другие формы – рецессивные. Иногда это заболевание возникает как результат аутоиммунных реакций организма.
При серьезных кровотечениях больным переливают плазму крови с достаточным содержанием белков, обеспечивающих свертываемость. Иногда используют препараты белков. В менее серьезных случаях назначают десмопрессин.
Серповидноклеточная анемия
Серповидноклеточная анемия – болезнь, которую многие помнят по школьному курсу биологии. С ее помощью иллюстрируют неполное доминирование гена.
У здорового человека эритроциты – это двояковогнутые диски. У больного серповидноклеточной анемией они принимают форму серпа из-за неправильного состава и, следовательно, формы гемоглобина.

Серповидные эритроциты не могут нормально переносить кислород, и это приводит к анемии. У человека с одной мутантной и одной нормальной копиями соответствующего гена эритроциты деформированы, но все еще переносят достаточно кислорода. В такие эритроциты хуже проникает малярийный плазмодий. Это одноклеточный паразит, возбудитель малярии – тяжелой болезни, распространенной в Индии, Африке и некоторых других странах. В этих же странах распространена и серповидноклеточная анемия.
Болезнь обычно проявляется в возрасте 3 месяцев. У младенцев отекают кисти и стопы, поскольку серповидные эритроциты забивают сосуды и мешают нормальному кровотоку. До 5 лет для детей с серповидноклеточной анемией очень опасны инфекционные заболевания – они более восприимчивы к сепсису, или заражению крови. Когда в организме накапливаются антитела, снижается и опасность для жизни.
У взрослых, страдающих серповидноклеточной анемией помимо собственно анемии, высокий риск закупорки сосудов почек и легких. Это приводит к нарушению работы этих органов и к смерти. Иногда эритроциты закупоривают сосуды глаз и вызывают слепоту.
Для лечения серповидноклеточной анемии применяют лекарства, которые либо препятствуют образованию неправильного гемоглобина, ответственной за деформацию эритроцитов, либо не дают эритроцитам застревать в капиллярах. Насколько эти препараты доступны нуждающимся – другой вопрос.
Прогерия
Прогерия – одна из самых редких болезней. Она развивается с 1,5-2 лет (детская прогерия) или с 20-30 лет (прогерия взрослых). По сути это преждевременное старение. Для прогерии характерны:
- Дряблость кожи и появление обесцвеченных участков и пигментных пятен;
- Атрофия мышц;
- Зубы и кости разрушаются;
- Волосы седеют и выпадают;
- Возникает атеросклероз.
При детской прогерии к 7–27 годам внутренние органы изнашиваются, и больной умирает. При прогерии взрослых средняя продолжительность жизни – 47–48 лет. Умственное развитие при прогериях обычно соответствует возрасту.

Изменения в организме, в том числе, хромосомные и клеточные, соответствуют нормальному старению. Ученые предполагают, что при прогерии их наступление ускоряется. Детская прогерия и прогерия взрослых вызваны мутациями разных генов, но все варианты прогерий рецессивны. Нужно две сломанные копии одного гена, чтобы болезнь проявилась.
Методы лечения прогерии в настоящее время в основном находятся в разработке. В 2020 году в США для лечения детской прогерии был допущен препарат лонафарниб, который препятствует правильному сворачиванию дефектных белков. Он продлевает жизнь больных примерно на 2 года. В остальном лечение остается симптоматическим.
Выводы и тезисы
К редким заболеваниям в России относят не только и не столько генетические болезни. В перечень орфанных заболеваний включены многие онкологические заболевания, нарушения обмена углеводов и минеральных веществ – железа, магния, фосфора, меди, туберозный склероз и другие. Большинство из них сложно либо диагностировать, либо лечить.
Отчасти причина этого – в малой изученности этих болезней. Другая причина – в том, что изучать и разрабатывать лекарства против орфанных заболеваний экономически невыгодно, поскольку они встречаются очень редко.
Тем не менее, для некоторых орфанных заболеваний разрабатываются методы лечения и диагностики. Современная генетика и биотехнологии значительно упрощают этот процесс.
- Орфанные заболевания – это редкие, зачастую наследственные болезни.
- В зависимости от государства и организации орфанными считаются разные болезни.
- Некоторые заболевания, редкие в одних странах, распространены в других.
- В России на февраль 2020 года орфанными считаются 262 болезни.
- Некоторые орфанные заболевания вызываются грибками, паразитами, бактериями или вирусами.
- Не все орфанные заболевания смертельны.
- Некоторые орфанные заболевания, вызванные нарушениями обмена веществ, можно корректировать с помощью диеты.
- Некоторые болезни могут быть полезны при определенных условиях.
- Орфанные заболевания редкие, поэтому не все они хорошо изучены.
Ответы на вопросы читателей
На титульном фото — Майкл и Эмбер Вандервит. Брат и сестра страдают от прогерии. Источник фото: https://www.thesun.ie/news/3639425/brother-and-sister-with-one-in-eight-million-benjamin-button-disease-reveal-heartbreaking-battle-against-cruel-bullies/
Подписывайтесь на наш канал в Telegram, чтобы получать свежие статьи своевременно!